Going Viral
Common Viruses May Trigger Autoimmune Diseases

Researchers think certain common viruses may trigger some autoimmune conditions—alone or in concert with other factors. A recent Office of Autoimmune Disease Research (OADR)-Office of Research on Women’s Health Science Talks series focused on understanding the triggers of autoimmunity and advancing research.
Almost 80 percent of people living with an autoimmune disease are women. It’s estimated there are 80-120 autoimmune diseases. These chronic and often debilitating diseases have no known cures. Some combination of genetics, immune regulation and the environment work together to form an “endotype” for each autoimmune disease patient, explained Dr. Judith James of the Oklahoma Medical Research Foundation.
Her presentation focused on lupus, or systemic lupus erythematosus (SLE), which disproportionately affects women. Nine women are diagnosed with SLE for every male. In SLE, the immune system attacks healthy tissue, causing inflammation and occasionally permanent damage.
Research from James’s lab has pinned Epstein-Barr Virus (EBV) as a potential trigger for SLE. More than 98% of adults in the U.S. have EBV antibodies, meaning they were infected with the virus at some point in their lives, most likely in childhood.
“How can an autoimmune disease be triggered by a nearly ubiquitous virus?” James asked. In other words, if EBV is so common in the general population, then why don’t more people develop SLE?
Her research focuses in part on the initial autoantibody response in patients with SLE. She learned that EBV infection is more prevalent in the SLE patient population. Their immune response of lupus patients targets different parts of EBV compared to healthy individuals.
She delved deeper by studying the autoantibodies, an antibody made against substances formed by a person’s own body, and their targets, called autoantigens, involved in the disease process of SLE. She discovered that the initial binding site on the autoantigen, named the epitope, is located in a peptide called Ro 169. She found that antibodies targeting this peptide cross-reacted with an EBV protein called EBNA1. This means antibodies could “confuse” the peptide for the EBV protein and mistakenly bind to Ro169 instead of EBNA1, potentially causing an autoimmune response. This phenomenon is known as molecular mimicry.

Her lab conducted numerous other studies that highlight the complex interplay between EBV, immune dysregulation and autoimmunity. Most recently her group showed this virus—which usually lays dormant in low numbers in healthy individuals—is more frequently reactivated in SLE patients. While there is still much to learn, her findings suggest that viral reactivation, molecular mimicry and functional mimicry may all contribute to developing SLE.
Dr. William Robinson, a professor of medicine at Stanford University Medical School, also researches the role of EBV in autoimmunity, but in a different condition: multiple sclerosis (MS). Similar to patients with SLE, almost all MS patients have been infected with EBV.
In MS, the immune system attacks myelin in the central nervous system. Myelin is the protective coating surrounding nerve fibers. When it is damaged, the immune system can also attack the nerve fibers within.
Robinson’s lab sought to understand the role of B cells in MS by sequencing the antibodies obtained from the spinal fluid of MS patients. He observed reactivity against several viruses, including EBV. Next, he narrowed his focus to one particular MS antibody that bound to EBV: MS39.
In another example of molecular mimicry, the MS39 antibody binds to both EBNA1 and to a molecule within the body: GlialCAM, a cellular adhesion molecule that is essential for maintaining the protective myelin sheath around nerve fibers. MS39 attacks both EBNA1 and GlialCAM, thus demyelinating the nerve fibers to possibly cause symptoms of MS.

Furthermore, Robinson observed a coordinated T cell response against both EBNA1 and MS39, suggesting that EBV-induced B cells may activate T cells that further contribute to the autoimmune response observed in MS.
“I see a critical role for EBV-mediated molecular mimicry with self-antigens among multiple human autoimmune diseases,” said Robinson.
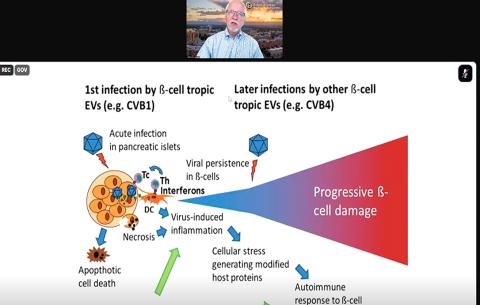
Dr. Marian Rewers shifted the discussion to a different chronic disease: type 1 diabetes (T1D). In many people with T1D, the immune system targets the pancreatic cells responsible for making insulin. Researchers have long suspected a link between viral infection and development of T1D but have not begun to make headway on this theory until recently.
A group of RNA viruses that may cause cold-like symptoms, called enteroviruses, have emerged as a prime suspect because of their ability to enter the pancreatic cells responsible for insulin production. These cells, known as islet cells, have a receptor called CAR (coxsackievirus and adenovirus receptor) that enteroviruses can utilize to infect the cell.
Enterovirus infection alone is not likely to cause T1D, said Rewers, professor of pediatrics and medicine, and executive director, Barbara Davis Center for Diabetes at the University of Colorado School of Medicine. Genetic predisposition also plays a role, combined with repeated enterovirus infections.
Rewers is a principal investigator for the TEDDY study (The Environmental Determinants of Diabetes in the Young). The study is a multicenter effort across Europe and the U.S. seeking to identify environmental triggers for T1D. The study has identified about 8,600 children as genetically high-risk for developing T1D and collects surveillance data regularly to monitor their health.
As of September 2024, 915 children had developed the “first step” of the pathway toward clinical diabetes, which Rewers defined as persistent islet autoimmunity marked by the presence of autoantibodies. Another 455 children had already progressed to clinical T1D.
TEDDY investigators conducted mass sequencing from patient stool samples and found that prolonged shedding of enterovirus B-type viruses was linked with the autoimmunity displayed in T1D. Intriguingly, they also learned that the autoimmunity displayed in T1D can be divided into two subtypes, which may determine when the disease begins to manifest.
Rewers thinks an enterovirus vaccine may be a useful preventative for individuals at high genetic risk for developing T1D, but this idea is still in the beginning stages.
TEDDY investigators have also found that recurrent rotavirus infections are linked to development of celiac autoantibodies in children at high risk for celiac disease, especially children on a high gluten diet.
“Everything from longitudinal cohort studies to applying single-cell technology to having good animal models” will be needed to further advance our understanding of the roles viruses play in autoimmune diseases, Robinson said. “We’re progressing overall, but there’s still work to be done.”